


(点此阅读原文)

拟南芥蛋白质组的质谱分析
期刊:Nature
时间:2020.03
2020年3月,Nature在线发表了来自德国慕尼黑工业大学Bernhard Kuster课题组合作题为“Mass-spectrometry-based draft of the Arabidopsis proteome”的研究论文。该研究首次发布了拟南芥的30个组织的转录组,蛋白质组和磷酸化蛋白质组的定量图谱。
研究背景
模式植物拟南芥彻底改变了我们对植物生物学的理解,并影响了生命科学的许多其他领域。拟南芥基因组在20年前被测序,此后在基因组和表观基因组水平上分析了数百个自然变异,相比之下,拟南芥蛋白质组作为大多数生物学过程的主要执行者,其特征还远远不够全面。
样本细则
30个不同组织:
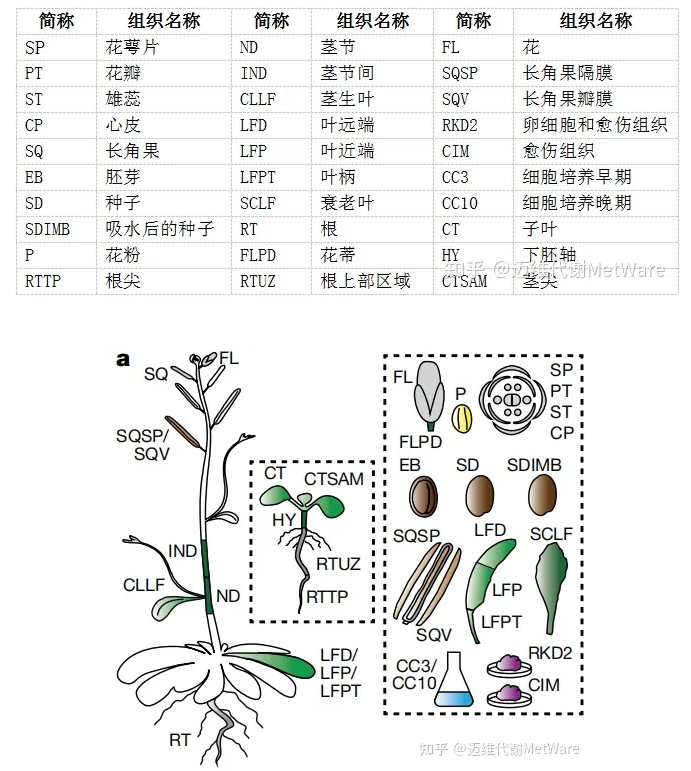
图1 组学所用样本
技术路线
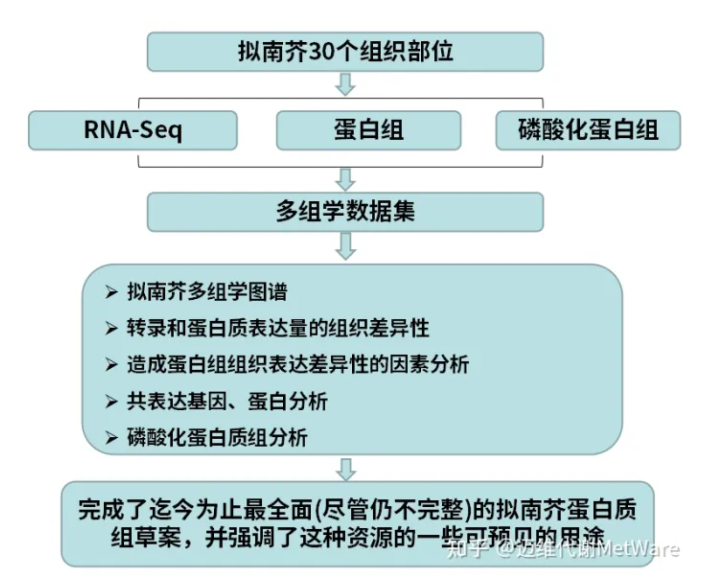
研究结果
#1
拟南芥多组学图谱
本研究基于可复制的生化和分析方法生成了一个多组学的表达图谱,平均每个组织17603±1317个转录本,14430±911个蛋白和14689±2509个磷酸化位点。拟南芥基因组Araport11注释的27655个蛋白编码基因中,18210个(66%)在本项目中有对应蛋白丰度的数据,与UniProt报告的蛋白编码基因比例(27%)相比,这是一个显著的增加,是早期组织蛋白质组分析中确定的蛋白质数量的两倍多(图3)。此外,本研究报告了43,903个磷酸化位点的定量证据,使该研究成为迄今为止发表的最全面的拟南芥磷酸化蛋白质组(图3)。
该图谱可以通过ATHENA(Arabidopsis THaliana ExpressioN Atlas; http://athena.proteomics.wzw.tum.de)和ProteomicsDB(http://www.proteomicsdb.org)网站进行分析。分析和可视化工具包括共表达分析、探索组织特异性的相互作用网络和预测肽序列的串联质谱的工具,该工具可用于验证已报道的肽鉴定(图4a)。ProteomicsDB进一步提供了超过328,000个未修饰肽段和超过43,000个磷酸化肽段的谱库,这些可以促进基于定量质谱的蛋白质分析的发展,这对拟南芥的研究特别有用,因为其抗体相对较少。
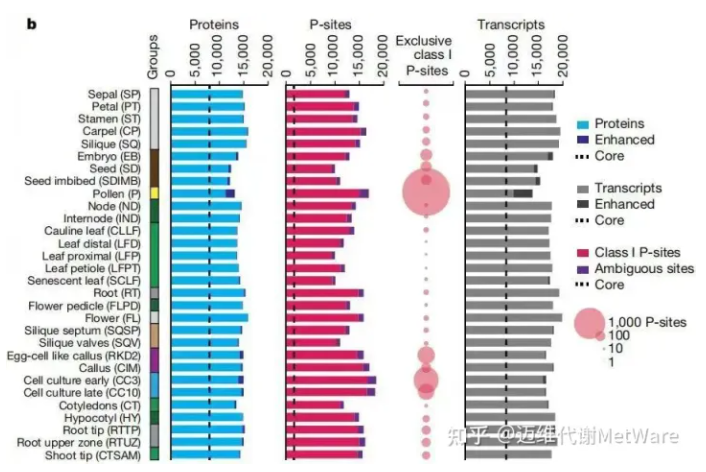
图2 不同组织得到的蛋白、蛋白磷酸化和转录水平的鉴定数
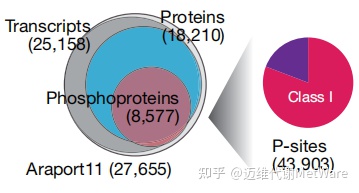
图3 与Araport11相比,转录组、蛋白质组、磷酸化蛋白组数据集中已鉴定的基因位点总数
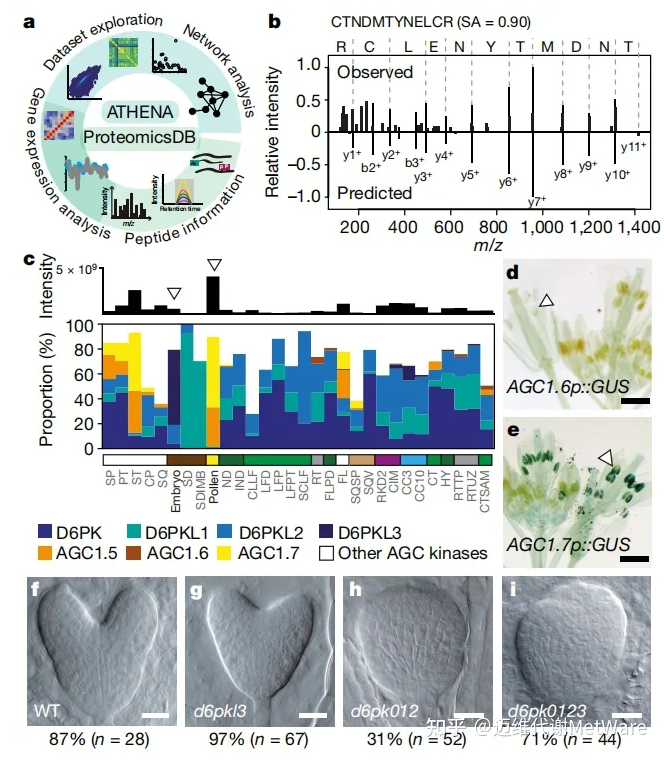
图4 ATHENA和ProteomicsDB中的数据探索
#2
蛋白组辅助基因组注释
利用Araport11,基于2776个N端肽段和2656个C端肽段的检测,蛋白质组数据证实了大量注释的开放阅读框边界。正如预期的那样,N端多肽通常表现出引发者蛋氨酸的裂解和N端乙酰化,这强烈依赖于与引发者蛋氨酸相邻的氨基酸。质谱数据平均覆盖每个蛋白序列的44%,使14115个蛋白亚型能够检测到独特的多肽。在297例中,数据还区分了2或3个剪接变体。对这些异构体特异性肽的选择使用合成肽进行了验证(确认率80%)。为了研究预测的短开放阅读框(sORF)的潜在翻译,本研究搜索了蛋白质组学数据与已发表和内部编译的sORF集合(ARA-PEP14;ATSO),共有51个不同的sORF被鉴定,随后使用合成肽进行了确认。这些结果表明了该图谱在完善拟南芥基因模型和蛋白质序列方面的潜力。
#3
蛋白表达量的组织差异性
蛋白质和转录本表达的动态范围分别跨越了6和4个数量级。如前所述,低丰度转录本的蛋白质证据被低估,这表明蛋白质组的覆盖还不够全面。低丰度转录本富集于基因本体(GO)数据库,如细胞信号传导或基因表达调控。尽管如此,蛋白质数据涵盖了大约50%的注释转录因子和75%的转录调控因子、激酶和磷酸酶,其中许多转录调控因子本身在翻译后被调节。值得注意的是,在蛋白质丰度的整个动态范围内检测到蛋白质磷酸化,这突出了所使用的磷酸肽富集的效率。
对单个组织或形态相似的组织(例如叶、种子或根)的分子数据进行分析,发现只有少数转录本或蛋白质以组织特异性的方式表达。相反,组织类型以不同的表达丰度模式为特征。例如,花器官表达了一组相似的基因,但在每个花组织里表达量是存在差异的。在这里,MADS-结构域转录因子显示出了预期的差异表达。
在蛋白质和转录水平上,组织之间的差异表达是非常明显的。值得注意的是,本研究发现在任何组织中,最丰富的蛋白质和mRNA在排序上几乎没有重叠,甚至没有一致性。研究检测到了种子的极端表达范围,例如,储存蛋白CRA1约占总蛋白的10%,十个最丰富的基因产物已经分别占总蛋白或转录本的30%以上。同样,RuBisCO复合物被称为地球上最丰富的植物蛋白,占总叶片蛋白含量的4-7% 。本研究注意到,尽管某些蛋白质在某些组织中占主导地位,但利用当前的技术可以对植物组织的蛋白质组进行非常深入的研究,反驳了长期以来认为植物蛋白质组学特别困难的观点。正如预期的那样,光合活性是组织间表达模式变化的主要因素之一,这也可能与质体定位蛋白及其丰度有关。然而,在转录和蛋白水平上,花粉的表达模式最为不同。
组织间表达模式的比较可用于确定单个蛋白的生物学功能,也可用于确定不同角度密切相关的蛋白家族成员的生物学功能。进一步研究AGCVIII激酶家族(拟南芥中有23个成员)发现,D6蛋白激酶(D6PK)亚家族成员在胚胎中含量高,AGC1.5和AGC1.7激酶成员蛋白在花和花粉中含量高(图4c)。与此结果一致的是,后续的实验显示AGC1.5和AGC1.7基因有启动子活性,但与AGC1.6密切相关的基因没有启动子活性,这有力地支持了前两个基因对花粉管生长的贡献(图4d, e)。本研究进一步揭示了D6PKs在胚胎发育中的重要作用,联合敲除可以协同增加功能缺失突变体中异常胚胎表型的发生(图4f-i)。
#4
决定不同组织中蛋白质含量差异的因素分析
一个特定基因翻译的蛋白质含量是由一个包括转录本的合成与分解、翻译和翻译后修饰的调控系统决定的。本研究发现在大多数组织中,转录本与蛋白水平正相关(Pearson 's correlation r = 0.28-0.7),大多数高或低丰度转录本分别产生高或低丰度蛋白(图5a)。为了进一步确定蛋白质丰度的分子决定因素,研究实施了一种模型选择方法,并测试了所选择的每个特征在组织特异性或全局水平上蛋白质水平变化的潜力。除了众所周知的预测因素,如转录水平和密码子使用,进化保守性、mRNA序列基序和蛋白质相互作用的数量是蛋白质水平的重要预测因子(图5b)。然而,仍有很大比例(48%)的蛋白质丰度变异未得到解释,这表明许多其他具有巨大个体效应但整体效应较低的分子因素可能在起作用,需要仔细的个案实验来解决。
蛋白质与转录本丰度图清楚地显示,同样丰富的转录本往往导致蛋白质丰度差异超过100倍。基因的蛋白- mRNA比(PTR)的这些差异可能是由于给定转录本的翻译效率、转录本稳定性和/或各自蛋白的稳定性的差异导致(图5c)。在参与光合作用或能量代谢的基因(如petA和rbcL)中检测到高PTRs,这表明这些大量需要的蛋白(和转录本)的翻译和稳定性得到了优化。相反,低PTRs的基因在诸如激素介导的信号通路等过程中富集,在植物中,这通常涉及蛋白质降解。生长素不稳定的AUX/IAA蛋白家族成员(如IAA8和IAA13)尽管转录水平高,但蛋白信号却很低,而且已知它们在生长素独立的蛋白酶体降解过程中受到严格控制(图5a)。
本研究还观察到PTRs的组织间变异,尤其是种子和花粉(图5d)。在种子中,低PTR的基因主要在蛋白质水平上表现出差异丰度,而蛋白质组在不同时间的检测结果表明,在萌发后,低PTR蛋白从储存的mRNA中快速翻译,以响应环己亚胺(CHX)(翻译模块)或MG132(蛋白酶体模块)处理。具有高PTRs的蛋白质则没有这种行为,这表明它们储存在种子中是为了便于萌发(图5e, f)。组织间波动的PTRs表明在转录或蛋白水平上有组织特异性调控。相比之下,具有稳定PTRs的基因在不同组织中可能受到类似的调控,因此拟南芥可利用的大量转录组资源可以用来估计相对蛋白质丰度。同样,磷酸化位点丰度与相应蛋白质水平之间的稳定比例表明,在稳态条件下,组织间磷酸化丰度的大多数变化可以归因于潜在蛋白质丰度的波动。
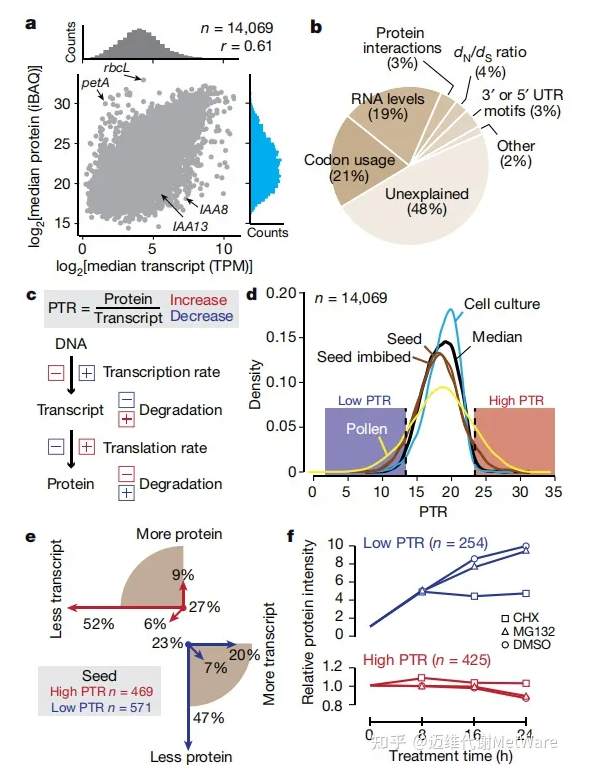
图5 蛋白质和mRNA的表达
#5
旁系同源和复合体的共表达分析
基于“关联推定”的思想,在转录或蛋白水平上,基因共表达分析被广泛用于确定基因之间的共同功能。本研究发现,共同表达的基因对也有很高的STRING得分,表明存在机体或功能上的相互作用。为了说明这一点,本研究专注于基因同源体和相互作用蛋白的共表达分析。与大多数植物相似,拟南芥经历了基因组复制,并在基因组中保留了相当数量的高度保守的旁系。与随机选择的基因对相比,重复基因表现出明显的正向表达相关性(包括转录和蛋白质)。基因冗余功能的这一指标得到的支持是,同源序列越丰富的敲除突变体越有可能显示可测量的植物表型。这种比较表达分析有助于对功能冗余基因的敲除组合进行优先排序。
AtPIN中记录的参与物理相互作用的蛋白在组织中的共表达分析表明,大约26%的AtPIN对可能是稳定的。正如预期的那样,基于蛋白质的相关性始终高于转录。因为共表达分析往往会产生大量相互作用的候选基因,本研究试图利用体积排阻色谱-质谱(SEC-MS)实验的数据作为独立的实验方法来优先研究这些基因。发现同时在组织图谱中共表达、SEC-MS中的共检测的候选基因数目会更少(图6a, b)。使用这种结合了来自体积排阻层析(SEC)和组织图谱数据集的信息的方法,对已知蛋白复合物的识别持续改进。
值得注意的是,在种子组织中,衣被蛋白复合体的ζ-亚基几乎完全由ζ-1同源提供,而在所有其他组织中几乎不存在这种亚基。亚单位丰度的比较也提供了复杂化学计量学的信息,当使用SEC-MS或组织图谱数据集时,也获得了稳定配合物的类似结果(图6c)。因此,本研究得到的组织图谱资源可以提供已知配合物相对亚基频率的初始近似值与未解决的化学计量。这对于很难进行生物化学研究的膜或细胞壁相关复合物特别有用。
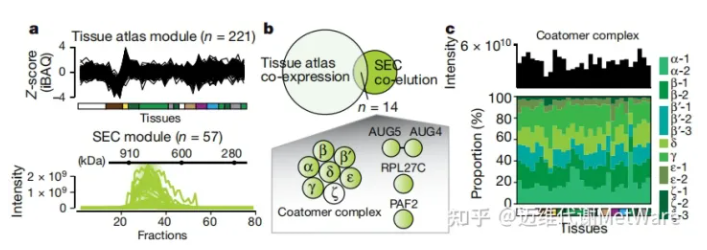
图 6 蛋白共表达和SEC-MS对蛋白复合物的表征
#6
拟南芥的磷酸化蛋白质组分析
在大多数组织中,检测到的激酶(642±55)和磷酸酶(119±6)的数量以及单个家族的比率是可比较的。然而,花粉和卵细胞样愈伤组织突出,这表明在这些组织中广泛的信号活动。与现有的磷酸化蛋白质组学数据相比,激酶-底物关系的信息仍然很少,这对于理解信号通路的级联或通路活性至关重要。由于这些关系很难通过实验发现,科学家们通常一开始采用计算方法。本研究使用motif- X算法确定了266个磷酸化motif,它们分为“脯氨酸定向”、“酸性”、“基本”或“其他”基序类别。结合组织中激酶和P-sites共发生的信息,以及外部共定位或相互作用信息,推测该数据集可以在解开激酶-底物和激酶- 磷酸化位点的关系上提供帮助。
总的来说,每个蛋白质的P位点数量表现出巨大的差异。例如,LEA蛋白家族在其序列中几乎每一个丝氨酸、苏氨酸或酪氨酸残基都被磷酸化(图7a)。这些参与种子成熟和干燥的非调控蛋白的磷酸化可能是调节其构象状态或相变的机制。相反,其他蛋白在调控区域显示了P-sites簇,包括受体样激酶的近膜区域,这表明它在招募互动伙伴方面的作用类似于人类受体酪氨酸激酶。
磷酸化事件的检测并不直接表明功能后果,但它确实提供了一个重要的起点。例如,脱落酸(ABA)受体RCAR10,参与ABA信号级联,被发现在四个位点磷酸化。本研究构建了磷酸化突变体(S113D和S32D),与野生型受体相比,它们对ABA的反应减弱(图7b)。Ser113是2C型蛋白磷酸酶(PP2C)结合表面的一部分,这种降低的反应可能与ABA结合或PP2C相互作用的影响有关。对于位于受体外周的Ser32,观察到的效应更可能是由于与其他调控蛋白的相互作用的改变。值得注意的是,两种RCAR10磷酸化突变体都改变了RCAR10 - pp2c共受体组合的ABA应答,这表明在发育和胁迫应答过程中,相互作用谱和基于磷酸化的ABA信号微调发生了变化。
作为蛋白质磷酸化功能后果的第二个例子,作者研究了QKY(也被称为MCTP15),它定位于胞间连丝,参与花和角果形态发生。因为它的一个p位点(Ser262)在所有检测的组织中都被检测到,并且在其他MCTP家族成员的第一个和第二个C2结构域之间发现了更多的p位点,因此推测磷酸化Ser262可能对QKY功能产生影响。为了验证这一假设,本研究在强QKY -9突变背景下产生了表达拟磷和磷酸化QKY转基因的植物株系。拟磷蛋白而非磷酸化蛋白构建拯救了花和角果的突变表型,这表明正常的QKY功能需要磷酸化Ser262(图7c-f)。
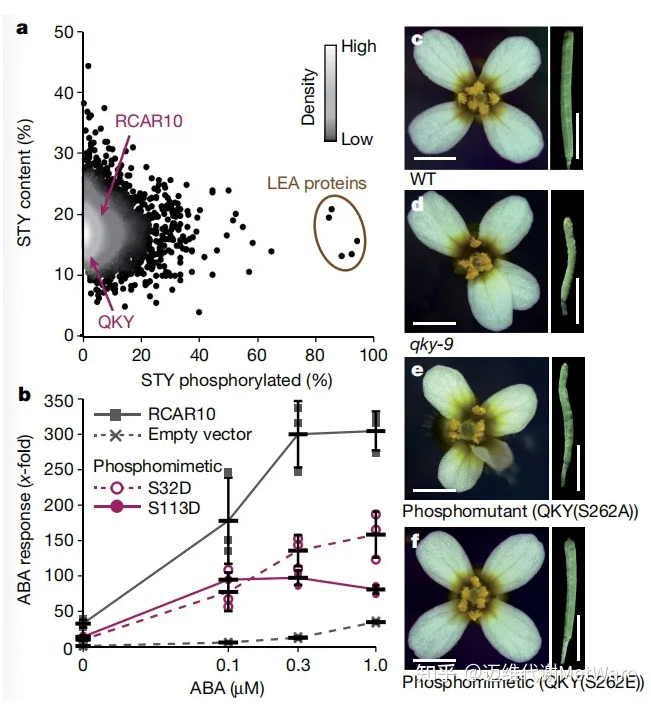
图7 蛋白质磷酸化功能分析
迈维小结
研究目的:构建拟南芥的不同组织蛋白表达图谱。
研究结论:本研究完成了迄今为止最全面(尽管仍不完整)的拟南芥蛋白质组草案,并强调了这种资源的一些可预见的用途。然而,仍有许多工作需要完成,包括更系统地覆盖蛋白质序列变异或翻译后修饰。数以万计的新发现的磷酸化位点等待功能鉴定,这是未来的一个特殊挑战。文中的例子表明,调查不同水平的组学数据可以导致对生物过程的新见解。因此,预计该资源以及提供的在线计算工具将使拟南芥研究社区能够执行多种类型的系统级分析。我们也期待这项研究将更广泛地影响植物研究,因为这项工作表明,基于质谱的定量蛋白质分析是克服植物研究缺乏抗体的一种方法。
值得借鉴:蛋白质组学是生命科学进入后基因组时代的必然产物和未来的重点研究方向。然而在大多数植物中,蛋白质的特征研究还远远不够全面,本研究基于不同组织进行蛋白质表达模式、相关性及修饰的研究值得借鉴。